Overview
This vignette presents a multi-sample workflow in STID
for comparing infection-associated niches across samples, conditions, or
ordered time points. Starting from single-sample niche objects, it
constructs separate comparative and temporal multi-sample objects and
evaluates how niche composition, spatial organization, molecular
programs, and pathogen distribution vary across samples.
This vignette covers:
- constructing multi-sample niche objects for comparative and temporal analyses;
- quantifying pathogen-positive burden, tissue and cell-type composition, and cellular aggregation;
- identifying niche-associated genes and cell–cell communication patterns across samples;
- inferring pathogen invasion trajectories and temporal gene modules;
- quantifying spatial organizational entropy during infection progression.
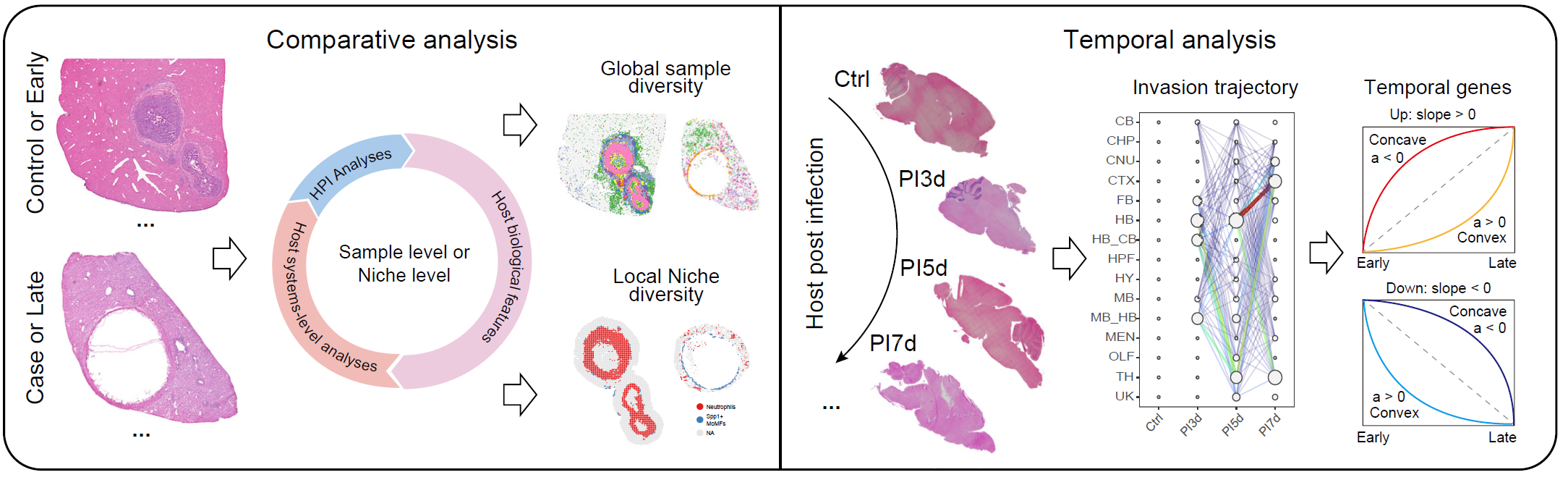
Overview of multi-sample niche analysis.
Note: Set the input
STIDobject, sample identifiers, comparison mode, niche keys, metadata keys, annotation columns, color vectors, gene filters, output group names, and figure paths according to the dataset being analyzed.
Reference code: For more runnable code examples, please refer to the article code.
Prerequisites
Note: The core workflow uses
tidyverse,Seurat, andSTID. Downstream sections may additionally requireCellChatandMfuzz.
Example data
Prepare a comparative multi-sample niche object
This example uses the single-sample niche object generated in the
previous vignette. Here, STID_obj_SS contains expanded
niche boundaries and associated metadata for the AE dataset.
CreateMultiSampNiche() aggregates selected single-sample
niches into a multi-sample object. In comparative mode,
loop_id specifies the samples or sample groups to
compare.
STID_obj_MS_comparative <- CreateMultiSampNiche(
STID_obj = STID_obj_SS,
multi_id = NULL,
loop_id = c("DPI_4_2", "DPI_79_1"),
compare_mode = "Comparative",
niche_key = "Niche",
description = NULL
)Note: Replace
STID_obj_SS,loop_id,compare_mode, andniche_keywith the corresponding object, sample identifiers, and niche key in your dataset. The downstream comparison identifier, such asComparative_2_4, must match an identifier stored in the resulting multi-sample object.
Prepare a temporal multi-sample niche object
This example uses the expanded focal-niche object generated by the
infection-associated niche identification workflow. Here,
STID_obj_aggregated contains expanded niche boundaries for
the JE dataset.
For temporal analysis, construct single-sample niche metadata for each time point and then combine the ordered samples into a temporal multi-sample object. The example creates separate pathogen-infected niche and host-responsive niche.
# Define cell-type colors
cell_palette <- c(
"#E41A1C", "#377EB8", "#000080", "#4DAF4A", "#984EA3",
"#FF7F00", "#FFFF33", "#A65628", "#F781BF", "#66C2A5",
"#8DA0CB", "#E78AC3", "#A6D854", "#FFD92F", "#E5C494"
)
cell_levels <- sort(unique(STID_obj_aggregated@meta.data$new_cell))
if (length(cell_levels) > length(cell_palette)) {
stop("Extend `cell_palette` so that each `new_cell` level has a unique color.")
}
col_lasso_cell <- setNames(
cell_palette[seq_along(cell_levels)],
cell_levels
)
col_lasso_cell2 <- c(
"Adipocytes" = "#E41A1C",
"Astrocytes" = "#377EB8",
"Dendritic cells" = "#000080",
"Endothelial cells" = "#4DAF4A",
"Epithelial cells" = "#984EA3",
"Fibroblasts" = "#FF7F00",
"Macrophages" = "#FFFF33",
"Microglia" = "#F781BF",
"Monocytes" = "#66C2A5",
"Neurons" = "#8DA0CB",
"NK cells" = "#E78AC3",
"Oligodendrocytes" = "#A6D854",
"T cells" = "#FFD92F"
)
# Construct single-sample niche metadata
STID_obj_SS_temporal <- CreateSingleSampNiche(
STID_obj = STID_obj_aggregated,
niche_key = "Niche_microbe",
meta_key = list("M2_NicheDetect_STS_STS_JEV_multisamp_microbe_region"),
ROI_type = "ROI",
pos_colnm = "ROI_label",
center_colnm = "ROI_center",
edge_colnm = "ROI_edge",
all_label_colnm = "All_ROI_label",
all_dist_colnm = "All_Dist2ROIcenter",
description = NULL
)
STID_obj_SS_temporal <- CreateSingleSampNiche(
STID_obj = STID_obj_SS_temporal,
niche_key = "Niche_host",
meta_key = list("M2_NicheDetect_STS_STS_JEV_multisamp_host_region"),
ROI_type = "ROI",
pos_colnm = "ROI_label",
center_colnm = "ROI_center",
edge_colnm = "ROI_edge",
all_label_colnm = "All_ROI_label",
all_dist_colnm = "All_Dist2ROIcenter",
description = NULL
)
STID_obj_SS_temporal <- AddSSNicheCells(
STID_obj = STID_obj_SS_temporal,
meta_key = "raw",
select_colnm = "new_tissue",
niche_key = "Niche_microbe"
)
STID_obj_SS_temporal <- AddSSNicheCells(
STID_obj = STID_obj_SS_temporal,
meta_key = "raw",
select_colnm = "new_tissue",
niche_key = "Niche_host"
)
# Construct multi-sample niche metadata
STID_obj_MS_temporal <- CreateMultiSampNiche(
STID_obj = STID_obj_SS_temporal,
multi_id = "Temporal_1_2_3",
loop_id = c("D0_1","D3_1", "D5_1", "D7_1"),
compare_mode = "Temporal",
niche_key = "Niche_microbe",
description = NULL
)
STID_obj_MS_temporal <- CreateMultiSampNiche(
STID_obj = STID_obj_MS_temporal,
multi_id = "Temporal_1_2_3",
loop_id = c("D0_1","D3_1", "D5_1", "D7_1"),
compare_mode = "Temporal",
niche_key = "Niche_host",
description = NULL
)Comparative multi-sample analysis
Most single-sample niche functions can be applied to multi-sample
objects by setting samp_mode = "MS" and specifying the
relevant multi-sample loop_id. This section uses
STID_obj_MS_comparative to compare composition,
aggregation, differential expression, and cell–cell communication across
samples.
Note: Set
loop_id,samp_grp_index,meta_key,niche_key,group_by, and color vectors for each comparison. UseLoopAllMultito summarize all multi-sample groups or a specific identifier such asComparative_2_4for one comparison.
Tissue composition, cell-type composition, and cell aggregation
AddMSNicheCells() appends selected metadata to the
multi-sample niche object. CalSampComp() compares
pathogen-positive proportions or annotation-level composition, and
CalSampCAI() evaluates local aggregation of annotated cell
populations.
col_lasso <- c(
"HsPCs" = "#E41A1C",
"Hepatocytes" = "grey95",
"Infla Heps" = "#4DAF4A",
"Fibroblasts" = "#984EA3",
"Cho/Spp1+ cells" = "#FFFF33",
"Spp1+ MoMFs" = "#FF7F00",
"MoKCs" = "#377EB8",
"Neutrophils" = "#F781BF",
"B/plasma cells" = "#A65628",
"Others" = "#8DA0CB"
)
col_lasso2 <- c(
"HsPCs" = "#E41A1C",
"Hepatocytes" = "#66C2A5",
"Infla Heps" = "#4DAF4A",
"Fibroblasts" = "#984EA3",
"Cho/Spp1+ cells" = "#FFFF33",
"Spp1+ MoMFs" = "#FF7F00",
"MoKCs" = "#377EB8",
"Neutrophils" = "#F781BF",
"B/plasma cells" = "#A65628",
"Others" = "#8DA0CB"
)
# Pathogen-positive proportion
STID_obj_MS_comparative <- AddMSNicheCells(
STID_obj = STID_obj_MS_comparative,
loop_id = "Comparative_2_4",
meta_key = "M1_SpotDetect_Gene_AE_correct_after_all_gene_white",
select_colnm = "Label_all_gene_nFeature(sum)",
niche_key = "Niche"
)
CalSampComp(
STID_obj = STID_obj_MS_comparative,
samp_mode = "MS",
loop_id = "LoopAllMulti",
samp_grp_index = TRUE,
meta_key = "M1_SpotDetect_Gene_AE_correct_after_all_gene_white",
niche_key = NULL,
group_by = "Label_all_gene_nFeature(sum)",
col = rev(c("#E41A1C", "#377EB8")),
return_data = FALSE
)
# Tissue and cell-type composition
CalSampComp(
STID_obj = STID_obj_MS_comparative,
samp_mode = "MS",
loop_id = "LoopAllMulti",
samp_grp_index = TRUE,
niche_key = NULL,
group_by = "anno",
col = col_lasso,
return_data = FALSE
)
# Cell aggregation index
ms_cai <- CalSampCAI(
STID_obj = STID_obj_MS_comparative,
samp_mode = "MS",
loop_id = "LoopAllMulti",
samp_grp_index = TRUE,
meta_key = NULL,
niche_key = "Niche",
group_by = "anno",
k_neighbors = 8,
min_agg_size = 10,
dist_thres = 1,
col = col_lasso
)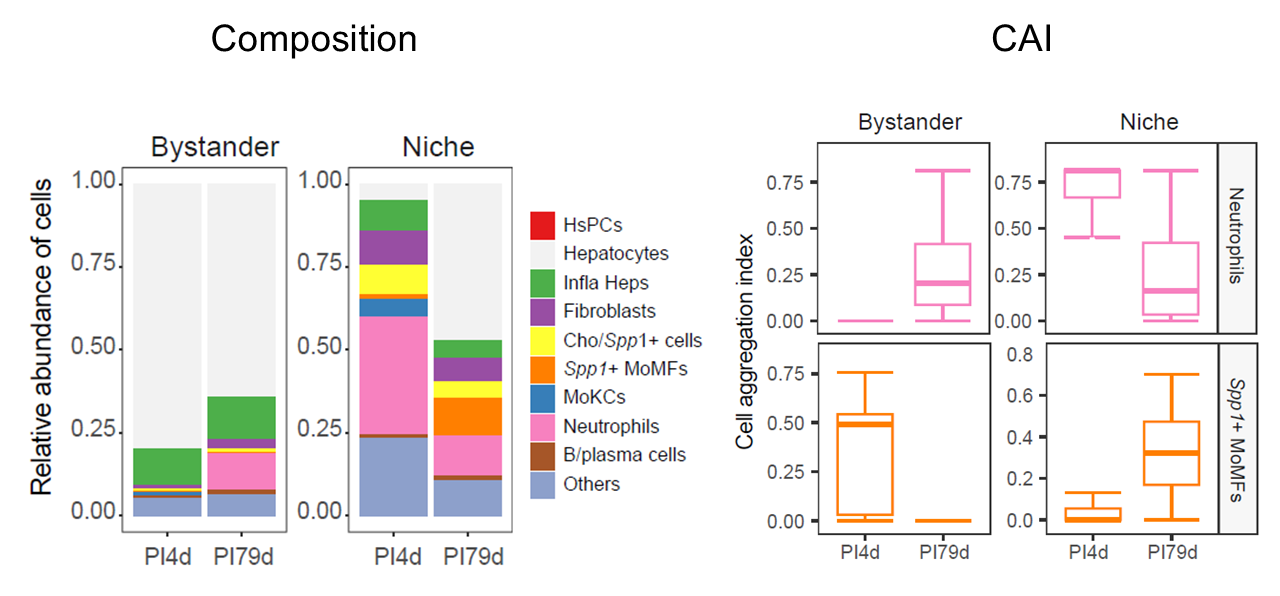
Pathogen-positive proportions, annotation composition, and aggregation across multi-sample niches.
Differential gene expression analysis
CalSampDEGs() identifies genes associated with selected
cell populations or niche groups across multi-sample comparisons. The
example filters predicted genes and pathogen-derived features before
testing host transcriptional differences.
Note: Set
loop_id,group_by,group_value,assay_id, thresholds, gene filters, andgrp_nmfor the comparison and feature space. If the selectedniche_keyyields too few genes, use broader annotation groups or adjust the tested group set.
MS_DEGs <- CalSampDEGs(
STID_obj = STID_obj_MS_comparative,
samp_mode = "MS",
loop_id = "Comparative_2_4",
samp_grp_index = TRUE,
logfc_thres = 2,
group_by = "anno",
group_value = c("Neutrophils", "Spp1+ MoMFs", "Fibroblasts", "B/plasma cells"),
assay_id = "Spatial",
padj_thres = 0.05,
adjust_method = "BH",
col = col_lasso,
remove_genes = c(
grep("^Gm", rownames(STID_obj_MS_comparative), value = TRUE),
grep("^EmuJ", rownames(STID_obj_MS_comparative), value = TRUE)
),
grp_nm = "Comparative_2_4_All"
)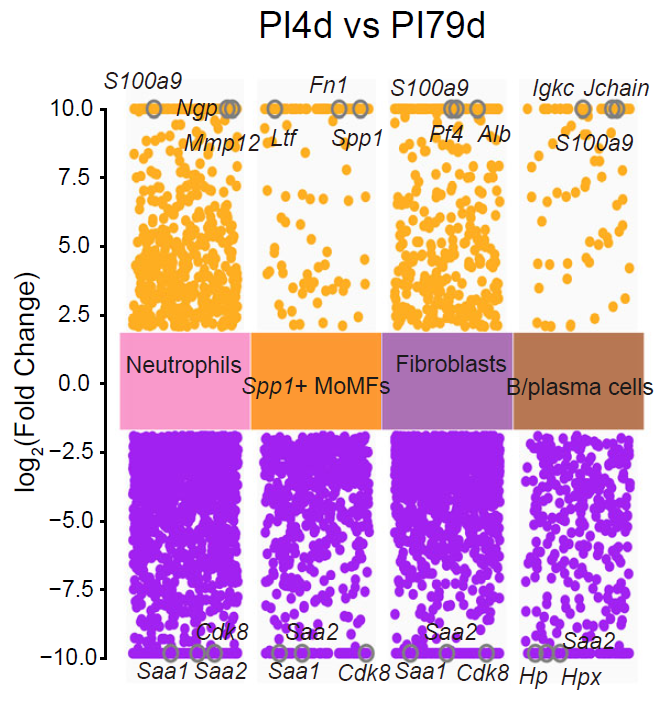
Differential expression analysis across comparative multi-sample niches.
Cell–cell communication analysis
Cell–cell communication results generated in the single-sample
workflow can be visualized in multi-sample niches by setting
samp_mode = "MS" in Plot_NicheCellComm(). This
reuses the same CellComm_data object for sample-level
comparisons.
Note: Use a
CellComm_dataobject generated from the same expression matrix and annotation scheme. Setloop_id, signaling pathways, ligand-receptor pairs, and color vectors according to the comparison.
CellComm_data <- readRDS("./outputdata/M3_CalSampCellComm/CalSampCellComm/CellComm_data.rds")
Plot_NicheCellComm(
STID_obj = STID_obj_MS_comparative,
CellComm_data = CellComm_data,
samp_mode = "MS",
loop_id = "Comparative_2_4",
signaling = c("CXCL", "CCL", "SAA", "SPP1", "MIF", "VEGF", "FGF"),
pairLR.use = NULL,
col = col_lasso2
)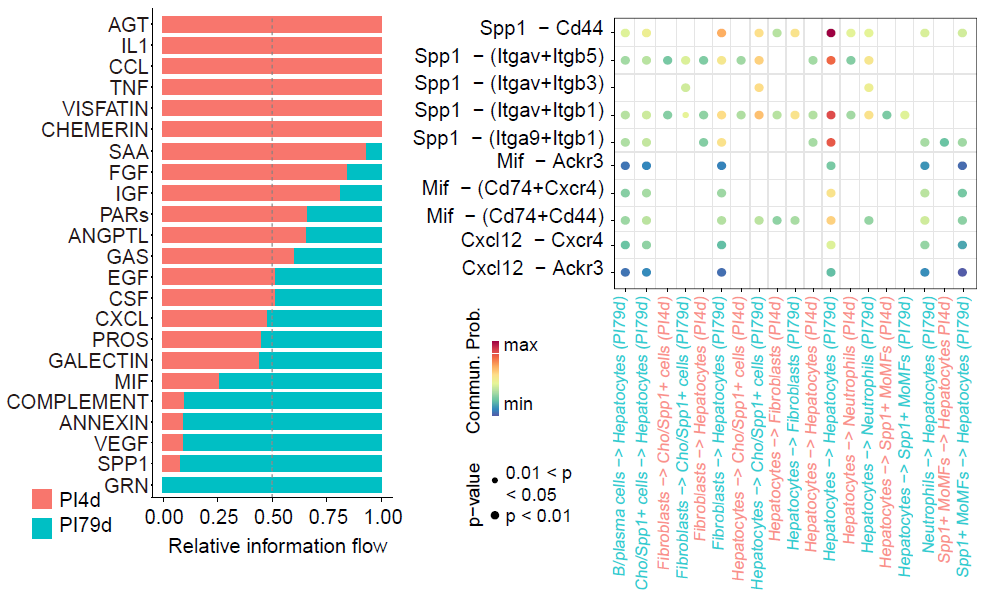
Cell–cell communication patterns across comparative multi-sample niches.
Temporal multi-sample analysis
Temporal multi-sample analysis uses ordered sample identifiers to
evaluate changes in pathogen distribution, spatial organization, and
host transcriptional programs. The following sections use
STID_obj_MS_temporal, which contains temporal niches
generated with compare_mode = "Temporal".
Note: Confirm that
loop_idfollows the biological time order, that metadata columns are harmonized across samples, and thatsamp_grp_indexmatches the structure of the temporal object.
Pathogen invasion trajectory analysis
CalSampPathoTrack() infers potential pathogen
propagation between adjacent time points by combining pathogen-positive
load in source annotations with the increase in pathogen-positive load
in target annotations.
For two adjacent time points, the propagation score is defined as:
Only target annotations with increased pathogen-positive load are retained. The resulting network summarizes the inferred direction and relative magnitude of pathogen spread across tissues or cell types.
Note: Set
loop_id,pos_colnm,neg_value,meta_key,group_by,col,grp_nm, anddir_nmto match the pathogen-detection metadata and annotation level. Use tissue-level or cell-type-level annotations according to the biological question.
CalSampPathoTrack(
STID_obj = STID_obj_MS_temporal,
loop_id = "Temporal_1_2_3",
pos_colnm = "Label_all_gene_nFeature(sum)",
neg_value = "neg",
samp_grp_index = FALSE,
meta_key = "M1_SpotDetect_Gene_JEV_multisamp_microbe_gene",
niche_key = NULL,
group_by = "new_cell",
col = col_lasso_cell,
return_data = FALSE,
grp_nm = "Temporal_1_2_3_cell",
dir_nm = "M4_CalSampPathoTrack"
)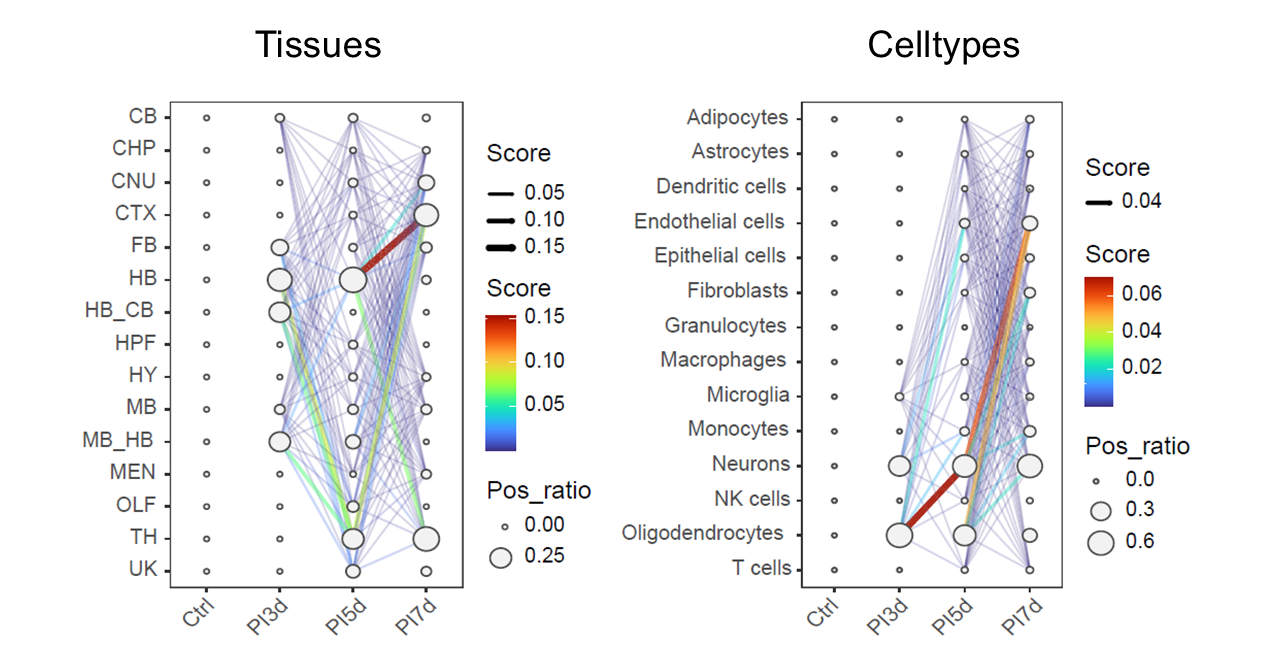
Inferred pathogen invasion trajectories across temporal multi-sample niches.
Spatial organizational entropy analysis
CalSampOSE() quantifies spatial organizational entropy
(OSE) to evaluate changes in local tissue or cell-type organization
during infection. Higher entropy indicates greater heterogeneity in
local spatial organization.
Spots are grouped into local spatial units, unit-type frequencies are summarized within each region, and entropy is normalized by the expected number of observed unit types to improve comparability across regions with different spot counts.
The adjusted entropy is:
where N is the total number of possible unit types and
n is the number of observed local spatial units.
Note: Set
loop_id,meta_key,group_by, color vectors,grp_nm, anddir_nmaccording to the temporal object. Useonly_plot = FALSEwhen entropy values are needed for downstream statistics.
CalSampOSE(
STID_obj = STID_obj_MS_temporal,
loop_id = "Temporal_1_2_3",
meta_key = "M1_SpotDetect_Gene_JEV_multisamp_microbe_gene",
group_by = "new_cell",
col = col_lasso_cell,
only_plot = TRUE,
return_data = FALSE,
grp_nm = "Temporal_1_2_3_cell",
dir_nm = "M4_CalSampOSE"
)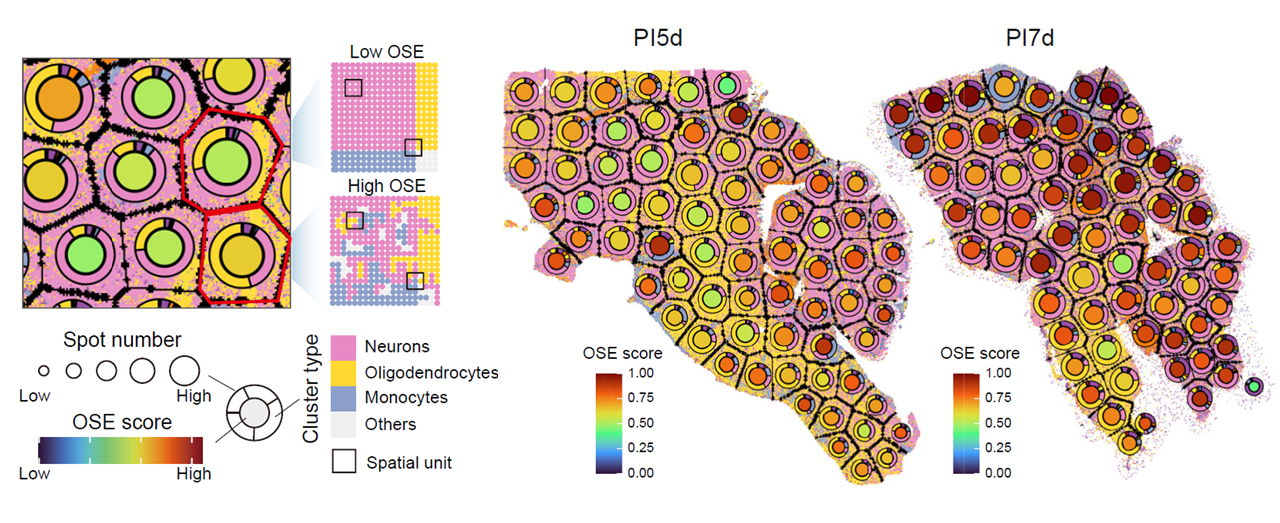
Spatial organizational entropy across temporal multi-sample niches.
Temporal gene module identification
CalSampGeneTrend() identifies dynamic host
transcriptional programs across ordered samples. The function supports
trend fitting for directional or nonlinear temporal patterns and fuzzy
clustering for module-level trajectories.
In fitting mode, genes are classified by linear and quadratic trend components. In clustering mode, fuzzy c-means clustering groups genes into temporal modules and identifies core genes by membership scores.
Note: Set
loop_id,meta_key,niche_key,group_by,gene_list,method, gene filters,grp_nm, anddir_nmaccording to the temporal comparison. Usemethod = "fitting"for interpretable trend classes andmethod = "mfuzz"for module-level trajectories.
# fitting
gene_trend_fit <- CalSampGeneTrend(
STID_obj = STID_obj_MS_temporal,
loop_id = "Temporal_1_2_3",
samp_grp_index = FALSE,
meta_key = "M1_SpotDetect_Gene_JEV_multisamp_microbe_gene",
niche_key = NULL,
group_by = NULL,
gene_list = NULL,
method = "fitting",
col = col_lasso_cell,
remove_genes = c(
grep("^Gm", rownames(STID_obj_MS_temporal), value = TRUE),
grep("Rik$", rownames(STID_obj_MS_temporal), value = TRUE)
),
return_data = TRUE,
grp_nm = "Temporal_1_2_3_fitting_all",
dir_nm = "M4_CalSampGeneTrend"
)
# mfuzz
gene_trend_mfuzz <- CalSampGeneTrend(
STID_obj = STID_obj_MS_temporal,
loop_id = "Temporal_1_2_3",
samp_grp_index = FALSE,
meta_key = "M1_SpotDetect_Gene_JEV_multisamp_microbe_gene",
niche_key = NULL,
group_by = NULL,
gene_list = NULL,
method = "mfuzz",
col = col_lasso_cell,
remove_genes = c(
grep("^Gm", rownames(STID_obj_MS_temporal), value = TRUE),
grep("Rik$", rownames(STID_obj_MS_temporal), value = TRUE)
),
return_data = TRUE,
grp_nm = "Temporal_1_2_3_mfuzz_all",
dir_nm = "M4_CalSampGeneTrend"
)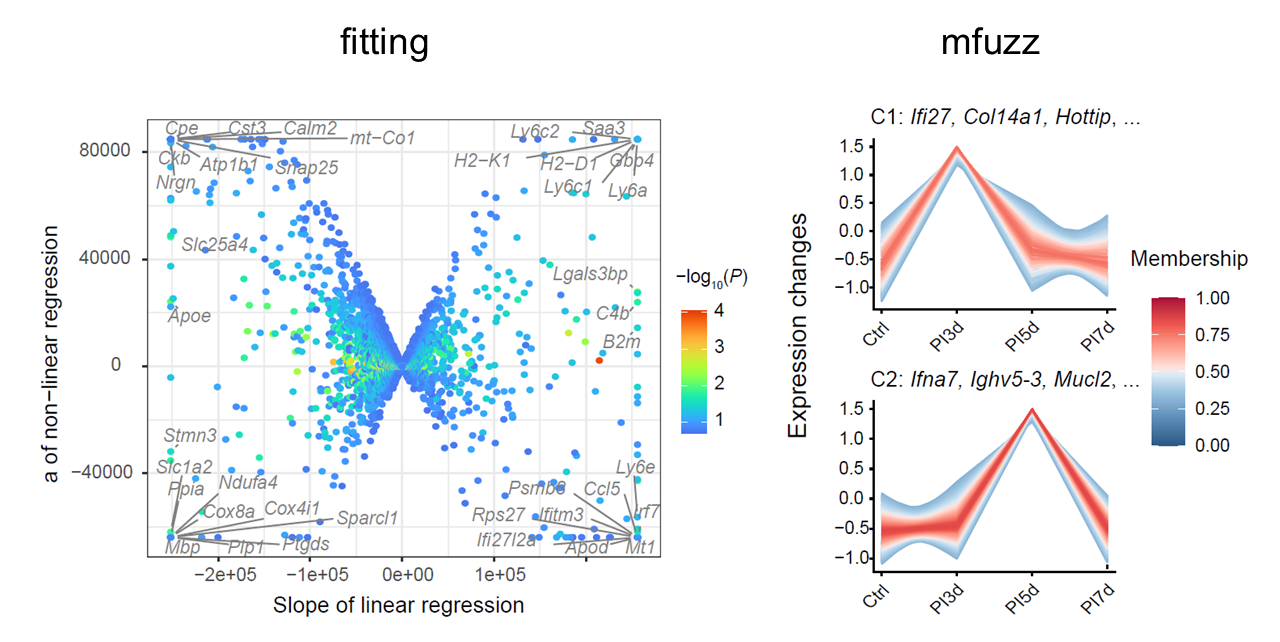
Temporal gene modules across ordered multi-sample niches.
Notes
- Confirm that sample identifiers, annotation columns, niche metadata, and named color vectors are consistent across all selected samples.
- Review
loop_id,compare_mode,niche_key,meta_key,group_by,samp_grp_index, and output names before running each section. - For temporal analyses, order
loop_idaccording to the biological time course and harmonize metadata levels across time points.
Next steps
This vignette demonstrates comparative and temporal analyses across
multiple samples to characterize niche remodeling, pathogen propagation,
spatial organization, and dynamic host responses during infection
progression. The generated results can be used for figure preparation,
biological interpretation, or customized downstream analyses with the
STID plotting
and utility
functions.
Session information
sessionInfo()
#> R version 4.2.0 (2022-04-22 ucrt)
#> Platform: x86_64-w64-mingw32/x64 (64-bit)
#> Running under: Windows 10 x64 (build 22000)
#>
#> Matrix products: default
#>
#> locale:
#> [1] LC_COLLATE=Chinese (Simplified)_China.utf8
#> [2] LC_CTYPE=Chinese (Simplified)_China.utf8
#> [3] LC_MONETARY=Chinese (Simplified)_China.utf8
#> [4] LC_NUMERIC=C
#> [5] LC_TIME=Chinese (Simplified)_China.utf8
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> loaded via a namespace (and not attached):
#> [1] digest_0.6.35 R6_2.6.1 jsonlite_1.8.8 lifecycle_1.0.5
#> [5] evaluate_1.0.1 cachem_1.1.0 rlang_1.1.7 cli_3.6.5
#> [9] rstudioapi_0.15.0 fs_1.6.3 jquerylib_0.1.4 bslib_0.8.0
#> [13] ragg_1.3.0 rmarkdown_2.29 pkgdown_2.2.0 textshaping_0.3.6
#> [17] desc_1.4.3 tools_4.2.0 htmlwidgets_1.6.4 yaml_2.3.10
#> [21] xfun_0.49 fastmap_1.2.0 compiler_4.2.0 systemfonts_1.0.4
#> [25] htmltools_0.5.8.1 knitr_1.49 sass_0.4.9